An Evolutionary-scale Model (ESM) for protein function prediction from amino acid sequences using the Gene Ontology (GO). Based on the ESM Cambrian Transformer architecture, pre-trained on UniRef, MGnify, and the Joint Genome Institute's database and fine-tuned on the AmiGO Boost protein function dataset, this model predicts the GO subgraph for a particular protein sequence - giving you insight into the molecular function, biological process, and location of the activity inside the cell.
"The Gene Ontology (GO) is a concept hierarchy that describes the biological function of genes and gene products at different levels of abstraction (Ashburner et al., 2000). It is a good model to describe the multi-faceted nature of protein function."
"GO is a directed acyclic graph. The nodes in this graph are functional descriptors (terms or classes) connected by relational ties between them (is_a, part_of, etc.). For example, terms 'protein binding activity' and 'binding activity' are related by an is_a relationship; however, the edge in the graph is often reversed to point from binding towards protein binding. This graph contains three subgraphs (subontologies): Molecular Function (MF), Biological Process (BP), and Cellular Component (CC), defined by their root nodes. Biologically, each subgraph represent a different aspect of the protein's function: what it does on a molecular level (MF), which biological processes it participates in (BP) and where in the cell it is located (CC)."
From CAFA 5 Protein Function Prediction
The following pretrained models are available on HuggingFace Hub.
| Name | Embedding Dim. | Attn. Heads | Encoder Layers | Context Length | QAT | Total Parameters |
|---|---|---|---|---|---|---|
| andrewdalpino/ESMC-300M-Protein-Function | 960 | 15 | 30 | 2048 | None | 361M |
| andrewdalpino/ESMC-300M-QAT-Protein-Function | 960 | 15 | 30 | 2048 | Int8W | 361M |
| andrewdalpino/ESMC-600M-Protein-Function | 1152 | 18 | 36 | 2048 | None | 644M |
| andrewdalpino/ESMC-600M-QAT-Protein-Function | 1152 | 18 | 36 | 2048 | Int8W | 644M |
First, install the esmc_function_classifier package using pip.
pip install esmc_function_classifierThen, we'll load the model weights from HuggingFace Hub, tokenize the amino acid sequence, and infer the GO terms.
import torch
from esm.tokenization import EsmSequenceTokenizer
from esmc_function_classifier.model import EsmcGoTermClassifier
model_name = "andrewdalpino/ESMC-300M-Protein-Function"
sequence = "MPPKGHKKTADGDFRPVNSAGNTIQAKQKYSIDDLLYPKSTIKNLAKETLPDDAIISKDALTAIQRAATLFVSYMASHGNASAEAGGRKKIT"
top_p = 0.5
tokenizer = EsmSequenceTokenizer()
model = EsmcGoTermClassifier.from_pretrained(model_name)
out = tokenizer(sequence, max_length=2048, truncation=True)
input_ids = torch.tensor(out["input_ids"], dtype=torch.int64)
go_term_probabilities = model.predict_terms(
input_ids, top_p=top_p
)You can also output the gene-ontology (GO) networkx subgraph for a given sequence like in the example below. You'll need an up-to-date gene ontology database that you can import using the obonet package.
pip install obonetimport networkx as nx
import obonet
# Visit https://geneontology.org/docs/download-ontology/ to download.
go_db_path = "./dataset/go-basic.obo"
graph = obonet.read_obo(go_db_path)
model.load_gene_ontology(graph)
subgraph, go_term_probabilities = model.predict_subgraph(
input_ids, top_p=top_p
)
json = nx.node_link_data(subgraph)
print(json)To quantize the model weights using int8 call the quantize_weights() method. Any model can be quantized, but we recommend one that has been quantization-aware trained (QAT) for the best performance. The group_size argument controls the granularity at which quantization scales are computed.
model.quantize_weights(group_size=64)You'll need the code in the repository to fine-tune and export your own models. To clone the repo onto your local machine enter the command like in the example below.
git clone https://github.com/andrewdalpino/ESMC-Function-ClassifierProject dependencies are specified in the requirements.txt file. You can install them with pip using the following command from the project root. We recommend using a virtual environment such as venv to keep package dependencies on your system tidy.
python -m venv ./.venv
source ./.venv/bin/activate
pip install -r requirements.txt
We'll be fine-tuning the pre-trained ESMC model with a multi-label binary classification head on the AmiGO Boost dataset of GO term-annotated protein sequences. To begin training with the default arguments, you can enter the command below.
python fine-tune.pyYou can change the base model and dataset subset like in the example below.
python fine-tune.py --base_model="esmc_600m" --dataset_subset="biological_process"You can also adjust the batch_size, gradient_accumulation_steps, and learning_rate like in the example below.
python fine-tune.py --batch_size=16 --gradient_accumulation_step=8 --learning_rate=5e-4Training checkpoints will be saved at the checkpoint_path location. You can change the location and the checkpoint_interval like in the example below.
python fine-tune.py --checkpoint_path="./checkpoints/biological-process-large.pt" --checkpoint_interval=3If you would like to resume training from a previous checkpoint, make sure to add the resume argument. Note that if the checkpoint path already exists, the file will be overwritten.
python fine-tune.py --checkpoint_path="./checkpoints/checkpoint.pt" --resumeTo simulate int4 quantized weights during training we can insert fake quantized tensors into the model and train like normal. The quantized model should perform better at inference time when some or all training epochs employ quantization-aware training.
python fine-tune.py --quantization_aware_training --quant_group_size=64 --resume| Argument | Default | Type | Description |
|---|---|---|---|
| --base_model | esmc_300m |
str | The base model name, choose from esmc_300m, esmc_600m. |
| --dataset_subset | "all" | str | The subset of the dataset to train on, choose from all, mf for molecular function, cc for cellular component, or bp for biological process. |
| --num_dataset_processes | 1 | int | The number of CPU processes to use to process and load samples. |
| --min_sequence_length | 1 | int | The minimum length of the input sequences. |
| --max_sequence_length | 2048 | int | The maximum length of the input sequences. |
| --unfreeze_last_k_layers | 7 | int | Fine-tune the last k layers of the pre-trained encoder network. |
| --quantization_aware_training | False | bool | Should we add fake quantized tensors to simulate quantized training? |
| --quant_group_size | 64 | int | The number of channels to group together when computing quantizations. |
| --batch_size | 8 | int | The number of samples to pass through the network at a time. |
| --gradient_accumulation_steps | 16 | int | The number of batches to pass through the network before updating the weights. |
| --max_gradient_norm | 1.0 | float | Clip gradients above this threshold norm before stepping. |
| --learning_rate | 5e-4 | float | The learning rate of the Adam optimizer. |
| --num_epochs | 50 | int | The number of epochs to train for. |
| --classifier_hidden_ratio | 1 | {1, 2, 4} | The ratio of hidden nodes to embedding dimensions in the classifier head. |
| --eval_interval | 2 | int | Evaluate the model after this many epochs on the testing set. |
| --checkpoint_interval | 2 | int | Save the model parameters to disk every this many epochs. |
| --checkpoint_path | "./checkpoints/checkpoint.pt" | string | The path to the training checkpoint. |
| --resume | False | bool | Should we resume training from the last checkpoint? |
| --run_dir_path | "./runs" | str | The path to the TensorBoard run directory for this training session. |
| --device | "cuda" | str | The device to run the computation on ("cuda", "cuda:1", "mps", "cpu", etc). |
| --seed | None | int | The seed for the random number generator. |
We use TensorBoard to capture and display training events such as loss and gradient norm updates. To launch the dashboard server run the following command from the terminal.
tensorboard --logdir=./runsWe can also infer the gene ontology subgraph of a particular sequence. The predict-subgraph.py script outputs a graphical representation of the predictions where green nodes have high probability and pink nodes have low probability.
python predict-subgraph.py --checkpoint_path="./checkpoints/checkpoint.pt" --top_p=0.1Checkpoint loaded successfully
Enter a sequence: MPNERLKWLMLFAAVALIACGSQTLAANPPDADQKGPVFLKEPTNRIDFSNSTG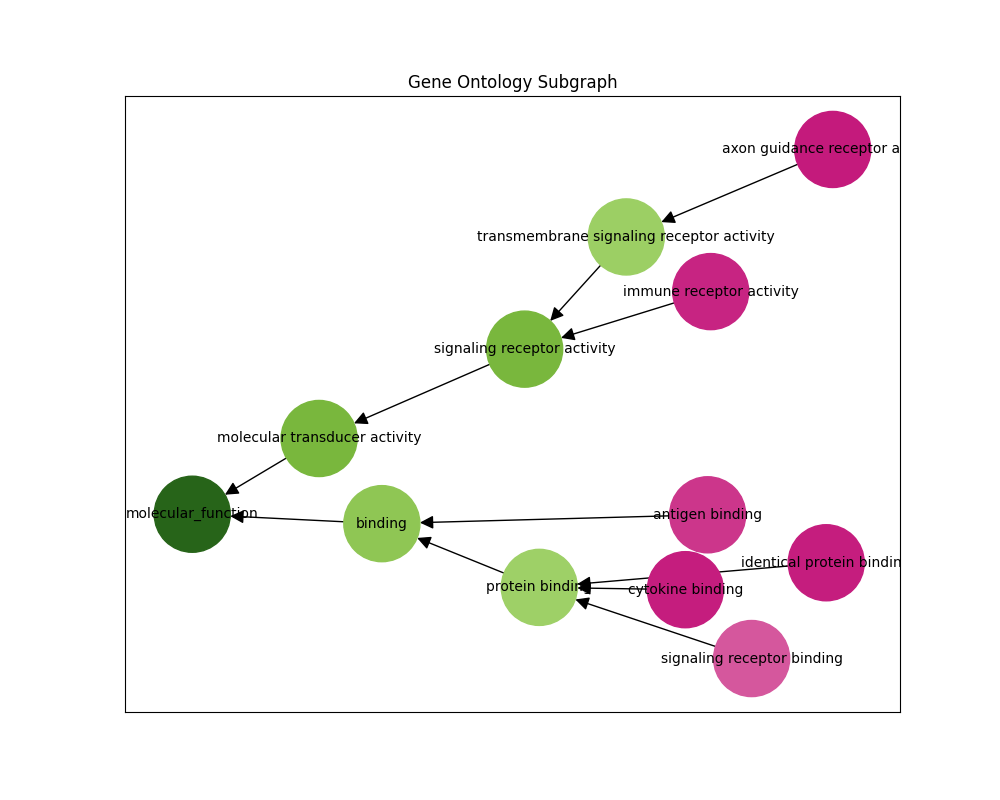
| Argument | Default | Type | Description |
|---|---|---|---|
| --checkpoint_path | "./checkpoints/checkpoint.pt" | str | The path to the training checkpoint. |
| --quantize_weights | False | bool | Should we quantize the weights of the model? |
| --quant_group_size | 64 | int | The number of channels to group together when computing quantizations. |
| --go_db_path | "./dataset/go-basic.obo" | str | The path to the Gene Ontology basic obo file. |
| --context_length | 2048 | int | The maximum length of the input sequences. |
| --top_p | 0.5 | float | Only display nodes with the top p probability. |
| --device | "cuda" | str | The device to run the computation on ("cuda", "cuda:1", "mps", "cpu", etc). |
| --seed | None | int | The seed for the random number generator. |
- T. Hayes, et al. Simulating 500 million years of evolution with a language model, 2024.
- M. Ashburner, et al. Gene Ontology: tool for the unification of biology, 2000.