
STONE (Determine RNA Structure with Truncation-mutation Signals within ONE Experiment), which uses feature engineering to integrate mutation and truncation signals from a single experiment, thereby enhancing signal analysis accuracy.
Create Environment with Anaconda python = 3.9.16 Python packages:
- pandas = 2.2.2
- numpy = 1.26.4
- scikit-learn = 1.4.2
- scipy = 1.13.0
- Matplotlib = 3.8.4
Software Requirement
- Fastp:https://github.com/OpenGene/fastp
- Bowtie2: http://bowtie-bio.sourceforge.net/bowtie2/index.shtml
- samtools: http://www.htslib.org/download/
- icSHAPE-pipe:https://github.com/lipan6461188/icSHAPE-pipe
- RNAFramework:https://github.com/dincarnato/RNAFramework
- Superfold:https://github.com/Weeks-UNC/Superfold
git clone https://github.com/sunlab/STONE.git
cd STONEInstall from the Package Archive File (.tar.gz):
install.packages("STONE/shapeTM_0.0.1.tar.gz", repos = NULL, type = "source")
library(shapeTM)The Rust software is compiled binary files that can be directly invoked on a computer or server with the same architecture.
[Click to view] Installation for different system architectures (not linux x86_64)
The entire software is built using the Rust language and managed with Cargo.
- Rust version:
rustc 1.79.0, cargo version:1.79.0 - Install Rust using
rustup:
Rust installation guide
-
Create a new Rust project:
cargo new project_name cd project_name -
Add the required dependencies:
cargo add memmap2=0.9.4 rayon=1.10.0 rand=0.8.5 clap=4.5.16
- Replace the contents of
src/main.rswith the provided.rsfile from the project.
-
Build the project:
cargo build --release
-
The binary will be located in the
target/release/directory.
- Use the countRT command from the icSHAPE-pipe software and convert the output file to a CSV file, resulting in a stop signal file.
- Use the rf-count command from the RNAFramework software to obtain a TXT file and an RC file. Then, use the rf-rctools command to convert the RC file into a CSV file, resulting in two mutation signal files. The "examples_script" folder contains sample bash scripts to obtain three sinal file.
example_data/ #Reference file needed for the script
├── example.sam
├── example.fa
└── example.len
RNAframework_folder/
├── example_output/ #This can be empty initially
├── tmp/ #Temporary folder used by the script
└── RNAframe_count.sh #The shell script
icSHAPE-pipe_folder/
├── example_output/
│ └── processed_files/ # Output files will be generated here
└── countRT.sh # The shell script
This pipeline utilizes the R package shapeTM to integrate three different signal files.
merge_outputs_auto(
path,
region = "human_small",
select_region = FALSE,
mini = FALSE
)
The subsequent analyses are performed using JupyterLab, and the associated Jupyter notebooks can be found in the stone_single_transcript_script/ directory.
- DMS Data Calculation: The DMS data is calculated using the notebook located at
stone_single_transcript_script/Single_transcript_DMS. - SHAPE Data Calculation: The SHAPE data is calculated using the notebook located at
stone_single_transcript_script/Single_transcript_SHAPE. - Raw Signals, STONE Structure Scores, and AUC Calculation: The pipeline also calculates the raw signals, STONE structure scores, and the AUC scores for each transcript.
This pipeline utilizes a RUST script located in stone_genome_software/ to integrate three different signal files.
(1) RNAframework's output
csv->[zip_rfcsv]->zippedcsv
txt->[zip_rftxt]->[zip_rftxt]->zippedtxt(2) icSHAPE-pipe's output
csv->[zip_pipe]->zippedpipe(3) Merge use the files in source above to generate mergedfile
zippedcsv----|
zippedpipe---|-> [merge] -> mergedfile ->
zippedtxt----|(4) extract or search exact all the genes in .bed file or search single gene through .bed file
mergedfile---> [bgsg] ----> all gene's result(1) zip_pipe Process the RT-stop count file produced by icSHAPE-pipe
Usage: {} <input_file> <output_file>
(2) zip_rfcsv: Compress the mutation count file generated by RNA Framework
Usage: zip_rfcsv --input <INPUT> --output <OUTPUT> --thread <THREAD> --strand <STRAND>
Options:
-i, --input <INPUT>
-o, --output <OUTPUT>
-t, --thread <THREAD>
-s, --strand <STRAND>
-h, --help Print help
-V, --version Print version
(3) zip_rftxt The mutation orientation file generated by the compressed RNA Framework
Usage: {} <input_file> <output_file> <num_threads>
(4) zip_rftxt2 The mutation directions were collated into the appropriate format
Usage: zip_rftxt --input <INPUT> --output <OUTPUT> --thread <THREAD> --strand <STRAND>
Options:
-i, --input <INPUT>
-o, --output <OUTPUT>
-t, --thread <THREAD>
-s, --strand <STRAND>
-h, --help Print help
-V, --version Print version
(5) merge merge three files upside
Usage: merge --csv <CSV> --pipe <PIPE> --txt <TXT> --output <OUTPUT>
Options:
-c, --csv <CSV>
-p, --pipe <PIPE>
-t, --txt <TXT>
-o, --output <OUTPUT>
-h, --help Print help
-V, --version Print version
(6) bgsg exact all genes from bedfile
Usage: bgsg --mergepath <MERGEPATH> --bedpath <BEDPATH> --output <OUTPUT>
Options:
-m, --mergepath <MERGEPATH>
-b, --bedpath <BEDPATH>
-o, --output <OUTPUT>
-h, --help Print help
-V, --version Print version
The subsequent analyses are performed using Python scripts, with the associated python script located in the stone_genome_software/ directory.
- Data Calculation: data is calculated using the notebook at
stone_genome_script/genome_model_output.py.
python genome_model_output.py -i /path/to/input_folder -o /path/to/output_folder -m /path/to/model.sav
This pipeline leverages superfold to fold RNA structures.
python2 /Superfold_1.0/Superfold.py output.shape --np 50
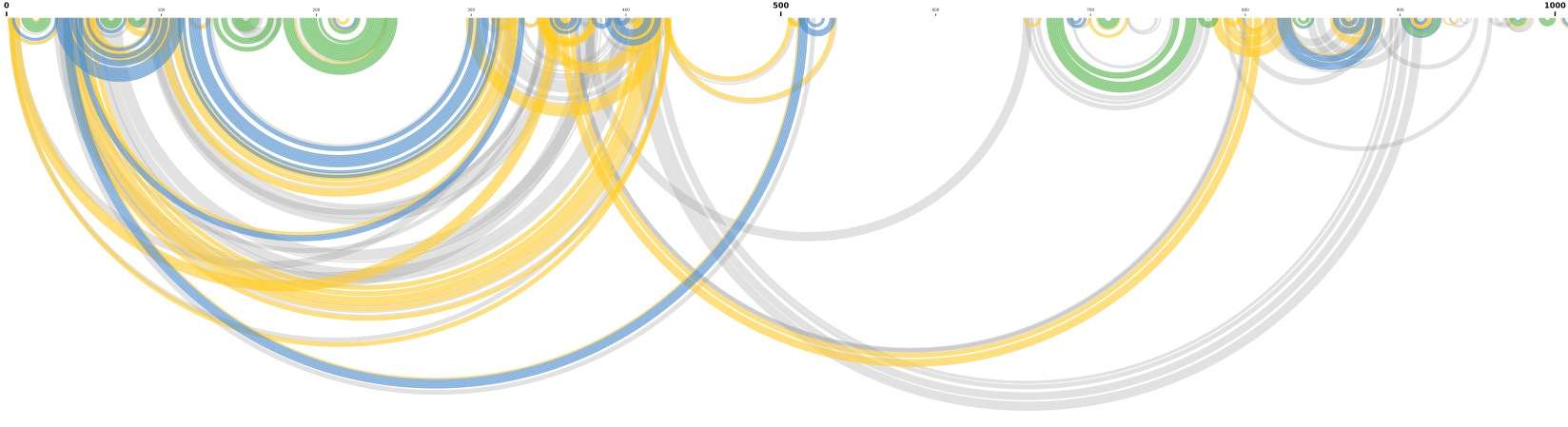
This pipeline employs a Python script named deltaSHAPE_stop_mut.py to identify RNA-binding protein (RBP) binding sites as part of an RNA structural exploration experiment.
python deltaSHAPE_stop_mut.py \
/path/to/model_output.csv \ # Path to the model output file
/path/to/stop_output.csv \ # Path to the stop mutation output file
/path/to/model_output.csv \ # Path to the model output file
/path/to/mut_output.csv \ # Path to the mutation output file
--mask5 10 \ # Number of nucleotides to mask 5' region
--mask3 10 \ # Number of nucleotides to mask 3' region
--pad 1 \ # Padding for the analysis
--Zcoeff 1.96 \ # Z-score coefficient
--Zthresh 0 \ # Z-score threshold for significant results
--SSthresh 1 \ # Secondary structure threshold for binding sites
--FindSite 2,3 \ # Find binding sites in specified regions
--out /path/to/result/output.txt \ # Output path for results
--pdf # Whether to generate a PDF plot
Options:
--mask5 <int>
Nucleotides to ignore at the 5' end of the sequence.
Default: `0`.
--mask3 <int>
Nucleotides to ignore at the 3' end of the sequence.
Default: `0`.
-p, --pad <int>
Smoothing window size (`2*pad + 1`).
Default: `1`.
-z, --Zcoeff <float>
Z-factor stringency coefficient.
Default: `1.96`.
-t, --Zthresh <float>
Z-factor threshold for significance.
Default: `0`.
-s, --SSthresh <float>
Standard score cutoff for significance.
Default: `1.0`.
-f, --FindSite <str>
Window size and minimum hits to identify binding sites (`pad, hits`).
Default: `'2,3'`.
-o, --out <str>
Output file location.
Default: `'differences.txt'`.
--magrank <store_true>
Sort output by deltaSHAPE values.
Default: `False`.
--all <store_true>
Include all nucleotides, with zero for insignificant changes.
Default: `False`.
--pdf <store_true>
Save the plot as a PDF.
Default: `False`.
--noshow <store_true>
Generate the plot but don't display it.
Default: `False`.
--noplot <store_true>
Skip the plot generation.
Default: `False`.
--dots <store_true>
Mark nucleotides passing Z-factor and standard score filtering.
Default: `False`.
--Zdots <store_true>
Mark nucleotides passing only Z-factor filtering.
Default: `False`.
--SSdots <store_true>
Mark nucleotides passing only standard score filtering.
Default: `False`.
--colorfill <store_true>
Highlight deltaSHAPE sites under the plot.
Default: `False`.
--ymin, --ymax, --xmin, --xmax <float>
Set plot axis limits.
Default: `-999` (auto).
-h, --help <store_true>
Display help message. We welcome contributions to the STONE project. To contribute, please fork the repository, make your changes, and submit a pull request.
- Issues: If you encounter any bugs or have suggestions, please open an issue on GitHub.
- Code Contribution: If you'd like to contribute code, please follow the steps outlined in our contribution guidelines.
- Testing: Ensure your changes are well-tested and update the documentation accordingly.
This project is licensed under the MIT License - see the LICENSE file for details.
If you use STONE in your research, please cite the following paper: