Cancer is a disease caused by progressive acumulation of genetic alterations either for single nucleotide variation (SNV) and/or copy number variation(CNV). There are some mutations important during initiation, promotion, and progression in cancer. However, it is important to identify these genetic alterations in cancer to understand the biology of the tumor as well as to identify genetic alterations that are of clinical importance either as a biomarker or as a therapeutic target.
This genetic alterations are detected using emerging technologies such as Next Generation Sequencing (NGS) with panels of cancer, exomes o complete genome.
The goal of this repository is show a workflow to analyze whole-exome sequencing (WES) using packages such as GATK4, mutect2, and CNVKit.
This is the general workflow.
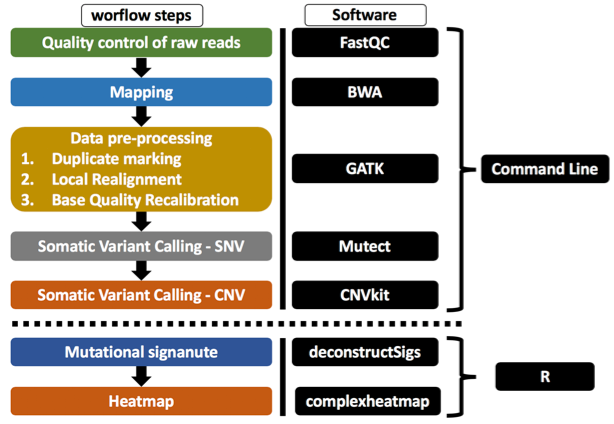
The repository is organized as follows
+-- workflow_to_analysis_WES
| +--bin/
| +-- 1_FastQC.sh
| +-- 2_Alignment.sh
| +-- 3_PreprocesingGATK4.sh
| +-- 4_Mutect2.sh
| +-- 5_CNVkit.sh
| +-- 6_MutationalSignature.rmd
| +-- 7_Heatmap.rmd
| +-- README.md
| +--data/
| +-- HeatMap
| +-- MutationalSignature
| +-- VariantsToStack
| +-- bam
| +-- fastq
| +-- sam
| +-- vcf
| +--figures/
| +-- Heatmap.pdf
| +-- SignatureCancer.jpeg
| +--Presentation.md
| +--README.md
| +--Repo_Abstract_and_Discussion.md
- bin: Contains the scripts to analyze WES and they are sorted as follows:
1_FastQC.sh - Contains scripts to evaluate the quality of the sequenced bases, the input file for this scrit are fastq file.
2_Alignment.sh - Contains script to align the readings using the bwa mem algorithm, the input file for this scritp are fastq file.
3_PreprocesingGATK4.sh - Contains script to preprocessing of data and generate a bam ready to variants calling, the input for this script is the output of 2_Alignment.sh or sam file.
4_Mutect2.sh - Contains scripts to call somatic single nucleotide variation (sSNV) using tumor only, the input for this script is the output of 3_PreprocesingGATK4.sh or bam file.
5_CNVkit.sh - Contains scripts to call somatic copy nucleótide variation (sCNV), the input for this script are bam file.
6_MutationalSignature.rmd - Contains scripts to analyze mutational signature, an example file used for this analysis is in Mutational Signature.
7_Heatmap.rmd - Contains scripts to plot variants and do heatmaps, an example file used for this analysis is in HeatMap.
- data: This directory contains different types of files that are used to execute the scripts.
- The fastq file contains data of breast cancer patients that were sequenced by the Ilumina HiSeq 2500 team, this file is the starting format for executing the first two script 1_FastQC.sh and 2_Alignment.sh.
- The sam file is the result of the 2_Alignment.sh that was generated from the fastq files and the input file for data 3_PreprocesingGATK4.sh.
- The bam file is the result of the 3_PreprocesingGATK4.sh and the input file for the so-called single-nucleotide variants and the number of copies.
- The vcf file is the file that is obtained after the call of bases using 4_Mutect2.sh and 5_CNVkit.sh, also contains the information of the genetic alterations found in these patients.
- The MutationalSignature file contains data to run 6_MutationalSignature.Rmd script and analyze mutagenic signatures to identify which mutagenic processes have operated in the life of these patients.
- The HeatMap file contains the data to run 7_Heatmap.Rmd and generate figures with the mutations found in these patients.
- figures: Figures performed in the R package
Before run these scripts, you need to download:
# For download Human GRCh37/hg19 move to data directory. Once in data make a directory
mkdir reference
cd reference
# Once in reference, execute the following command.
wget ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/hg19/ucsc.hg19.fasta.gz
Line command
ii) BWA
iii) GATK4, this package contains Mutect2
iv) CNVkit
R
vi) ComplexHeatmap