West Nile virus (WNV), a member of the Flavivirus genus, is transmitted in an enzootic cycle involving birds as amplifying hosts and mosquitoes as vectors, which can ultimately be transmitted to mammals, considered dead-end hosts, causing disease outbreaks in horses and/or humans. Currently, the virus is considered a recurrent zoonosis with a wide geographic distribution. Phylogenetically, WNV is classified into eight lineages. The Andalusian viral samples belonged to lineage 1 and were relatively similar to those of previous outbreaks which occurred in the Mediterranean region. A phylogenetic analysis was performed on the obtained consensus genomes in the context of a world-wide representative set of WNVs.
- download the sequences
- perform phylogenetic analysis
- document the work in GitHub
- prepare a presentation for the same
The first thing we did was create a working directory where we will save our files. we initiated a git repo this way we could share our work on GitHub
mkdir WNV
cd WNV
git inityou can copy this github repo using git clone in your terminal
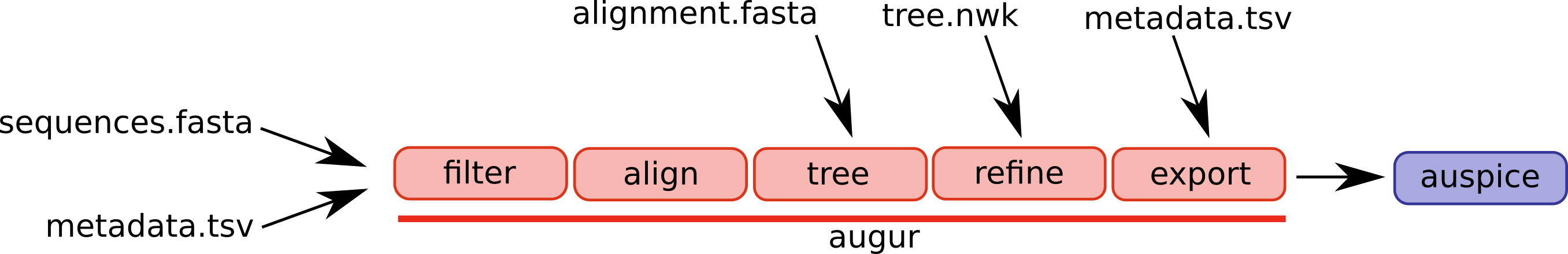
git clone git@github.com:Mattcreates25/West_nile_virus.gitAugur is a bioinformatics tool for phylogenetic analysis. the collection of commands from the tool are designed to be used with a larger processing pipeline like
snakemake Augur is composed of a series of modules and different workflows will use different parts of the pipeline.
A selection of augur modules and different possible entry points are illustrated below.
For Augur to run we created a unique environment where we could install the package using miniconda
conda create -n forAugur
conda activate forAugur
conda install -c conda-forge -c bioconda augurNextstrain’s auspice is an open-source interactive tool for visualizing phylogenetic data
conda install -c conda-forge nodejs
npm install --global auspicesnakemake is the pipeline tool preferred by Nextstrain in simplifying augur commands
conda install snakemake -c <channel>If you want to run the Snakefile use the commands found in the snakemake.md
snakemake
snakemake --cores 1 results/all_alignment.fasta
snakemake --cores 1 results/tree.nwk
snakemake --cores 1 results/branches.jsonoutput
Building DAG of jobs...
Using shell: /bin/bash
Provided cores: 1 (use --cores to define parallelism)
Rules claiming more threads will be scaled down.
Job counts:
count jobs
1 refine
1
[Fri Sep 2 10:43:00 2022]
rule refine:
input: results/tree.nwk, results/all_alignment.fasta, results/newmeta.csv
output: results/new_tree.nwk, results/branches.json
jobid: 0A message similar to this one is generated for each command run
download the sequences with wget
wget https://www.ebi.ac.uk/ena/browser/api/fasta/OU953897.1?download=true
wget https://www.ebi.ac.uk/ena/browser/api/fasta/OU953898.1?download=true
wget https://www.ebi.ac.uk/ena/browser/api/fasta/OU953895.1?download=true
wget https://www.ebi.ac.uk/ena/browser/api/fasta/OU953896.1?download=trueThe reference sequence was downloaded from NCBI and saved into a file called refseq.fasta
[1][https://www.ncbi.nlm.nih.gov/nuccore/NC_009942.1]
The fasta sequences for the worldwide representative set of WNVs sequences were obtained using batch entrez and imported into a single file called
WWR_sequences.fasta
accession numbers were retrieved from the viruses-13-00836-s001.zip which can be found in the Table S1.R3.xlsx. for retrieving purposes,
they were saved into a text file called WWrep_accession.txt this text file was then uploaded into batch entrez.
combine the four sequences into a file that contains all the sequences then combined them with the WWR_sequences.fasta
cat OU* >> four_sequences.fasta
cat WWR_sequences.fasta four_sequences.fasta >> all_sequences.fastawget https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8148183/bin/viruses-13-00836-s001.zip
unzip viruses-13-00836-s--1.zipcreate a txt file and paste all the accession numbers there. this file will be input into batch entrez
touch WWrep_accession.txtcreate a directory for all sequence data and move all the sequence data into that directory
mkdir data
mv *.fasta dataThe entire analysis can be run using snakemake workflow management. Snakemake breaks a workflow into a set of rules that are specified
in a file called Snakefile. Each rule takes a number of input files, specifies a few parameters, and produces output files
Parse delimited fields from FASTA sequence names into a TSV and FASTA file using a Snakefile input in this code creates a metadata file
alongside the fasta file and saved into a folder called results which will be used in subsequent steps
to do this we created a Snakemake file and used the augur parse command
rule parse:
input:
sequences = "data/all_sequences.fasta"
output:
sequences = "results/all_sequencesP.fasta",
metadata = "results/all_metadata.csv"
params:
fields = "strain"
shell:
"""
augur parse \
--sequences {input.sequences} \
--fields {params.fields} \
--output-sequences {output.sequences} \
--output-metadata {output.metadata}
"""
we Created a python script to split the accessions from the description in the metadata and mark the script as an executable file
# import the panda library
import pandas as pd
#read in the metadata csv
df = pd.read_csv('results/all_metadata.csv')
#check the format
df.head(15)
#create a variable with split data
new_metadata = df['strain'].str.split('.', n=1, expand=True)
#rename the columns
new_meta = new_metadata.rename(columns={0:'strain',1:'name'})
#export as a csv
new_meta.to_csv('newmeta.csv',index=False)marking the parse script as an executable file and run it using python parse.py in bash
touch parse.py
chmod +x parse.pyperform nucleotide base count using augur index
augur index -s data/all_sequences.fasta -o results/sequence_index.tsvWe then provide the sequence index as an input to augur filter commands to help in the filtering of sequence-specific attributes. encountered a problem here cause of a conflict in metadata opted to skip the filter option and worked with the entire data set
augur filter --sequences data/all_sequences.fasta --metadata results/newmeta.csv --sequence-index results/sequence_index.tsv --output filtered.fasta 310 strains were dropped during filtering
155 had no metadata
155 had no sequence data
ERROR: All samples have been dropped! Check filter rules and metadata file format.
for alignment mafft is required. MAFFT (Multiple Alignment using Fast Fourier Transform) and it is a high-speed multiple sequence alignment program.
sudo apt install mafft iqtree raxml fasttree vcftoolsaugur align -s data/all_sequences.fasta -o results/all_alignment.fasta --method mafft --reference-sequence data/refseq.fasta --fill-gaps
The phylogenetic tree was recovered by maximum likelihood, using a general time reversible model Augur version 14 + introduced --tree-builder-args which allows the user to create a tree with bootstrap values.
augur tree -a results/all_alignment.fasta -o results/tree.nwk --method iqtree \
--substitution-model GTR -o results/tree.nwk --tree-builder-args="-ninit 2 -n 2 -me 0.05"Branching date estimation was carried out with the least square dating (LSD2) method which is the default.
augur refine --tree results/tree.nwk -a results/all_alignment.fasta --metadata results/newmeta.csv \
--output-tree results/new_tree.nwk --output-node-data results/branches.json --keep-rootaugur export v2 -t results/new_tree.nwk --node-data results/branches.json --output results/branches_na.json \
--title 'West Nile Virus Outbreak in Andalusia' --maintainers 'mark https://github.com/Mattcreates25' \
--panels tree --metadata results/newmeta.csv --skip-validation
To view the tree we will have to write a narrative.md file that works alongside the dataset directory. This can be found in; narratives
---
title: west_Nile_narrative
authors: "Mark Njama"
authorLinks: "https://github.com/Mattcreates25"
affiliations: "icipe"
date: "August 2022"
dataset: "http://localhost:4000/results/branches/na?d=tree"
abstract: "This narrative will take us to auspice for vizualization."
---
after this you can view your tree by running this line in bash this creates a localhost with your tree(click on the localhost 4000 link)
auspice view --datasetDir results/ --narrativeDir narrative/
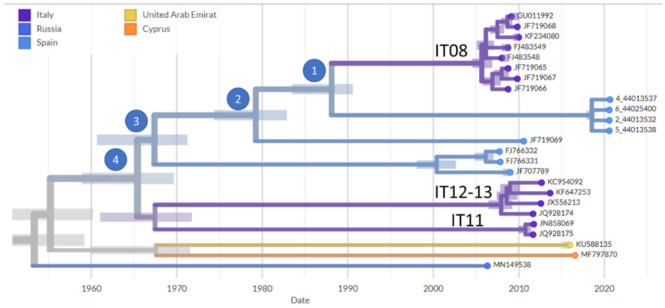
Sequences of the Spanish 2020 WNV outbreak OU the closest relatives from previous outbreaks in Italy and the sequence JF719069 from a lethal equine case in Andalusia (Spain) in 2010. Other Spanish outbreaks were: JF707789, from a mosquito in Huelva, FJ766331 and FJ766332 from a golden eagle in Toledo. Other related outbreaks from the Mediterranean region (Cyprus MF797870), or adjacent locations (United Arab Emirates KU588135 and Russia MN149538) are also included.
calculate non-synonymous to synonymous ratios along the viral genomes using the KaKs_Calculator
and find the The variability along the viral genomes using the Shannon entropy(basically we were measuring the uncertainty of the nucleotide distribution)
create a python script for the shannon entropy remember to install scipy
pip install scipy
touch shannon.py
chmod +x shannon.pyrun the python script shannon.py using python shannon.py
output
0.5032instead of using the kakscalculator tool with bash. we opted to Using R on the commandline we created a script called rscript.R and marked it as an executable file using chmod +x rscript.R
to run the script simply call Rscript rscript.R
output error
warning message:
In kaks(all_alignment, verbose = F, debug = F, forceUpperCase = T, :
sequence lengths are not a multiple of 3Due to this error we opted to use mega to calculated the non-synonymous to synonymous ratios along the viral genomes
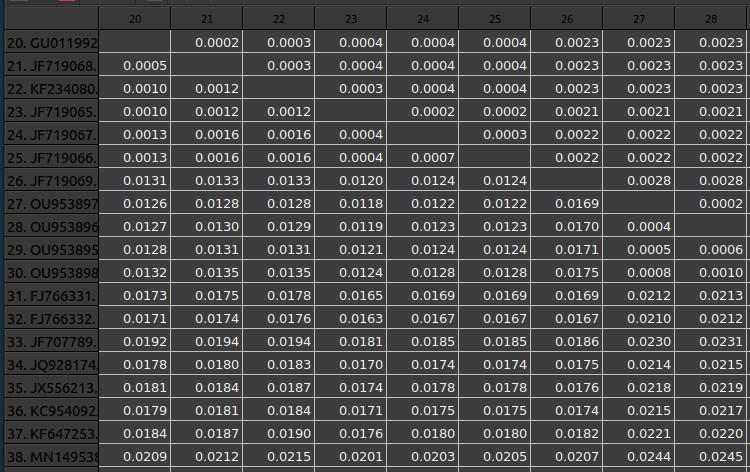
The phylogenetic analysis confirms the initial assignment to lineage 1. The table provides the estimated genetic distances among the WNV genome sequences, The values are average distances between all the sequences in the cluster and the compared sequence. the sequences GU011992, JF719068, KF234080, FJ483549, FJ483548, JF719065, JF719067, and JF719066, from the 2008–2009 Italian outbreak. additionally, the sequences KC954092, KF647253, JX556213, and JQ928174 from the 2012 and 2013 Italian outbreaks. the 3 Sequences JN858069 and JQ928175 are from the 2011 Italian outbreak.