Pedigree data in sgkit #786
Replies: 10 comments 4 replies
-
|
Thanks for starting this discussion @timothymillar! Somewhat related to pedigree is the notion of whether an allele is ancestral vs. derived, discussed at https://github.com/pystatgen/sgkit/discussions/580. I'm linking here just to remind myself of |
Beta Was this translation helpful? Give feedback.
-
|
For general graph wrangling we've had good luck with NetworkX (cf. https://github.com/related-sciences/nxontology). I think that's a reasonable dependency for us to pick up, as it's just Python. The alternatives, igraph and graph-tool, use C/C++ to improve performance but also complicate build and release. |
Beta Was this translation helpful? Give feedback.
-
|
I'm very positive about including pedigree support in sgkit @timothymillar . We've recently added support for pedigrees in tskit, and are currently working on methods to run popgen simulations conditioned on a given pedigree in msprime. Having support for a sensible pedigree representation and conversion utilities from things like (the various forms of) PED would be very helpful when processing pedigrees. The approach we've taken is to encode pedigree data using the "individual table", where each row corresponds to the information for a particular individual and individuals are referred to by their zero-based integer IDs (very much in the spirit of sgkit). For example, here's a simple simulated Wright Fisher pedigree in an individual table, and the corresponding graph (produced using networkx):
(Ignore the flags and location columns, these aren't important). The way we've encoded the pedigree information is to add a "parents" column to the table. The parents column/array can have any dimension, making things very flexible (actually the column is ragged, so that one row can have 0 parents and others have any number). We found that fixing on a (N, 2) array was more convenient for most purposes. The ID -1 here is used as NULL or "unknown". What I'd suggest for sgkit pedigree data would be to have something similar. We could have a dataset which at a minimum expects there to be a (N, k) I think |
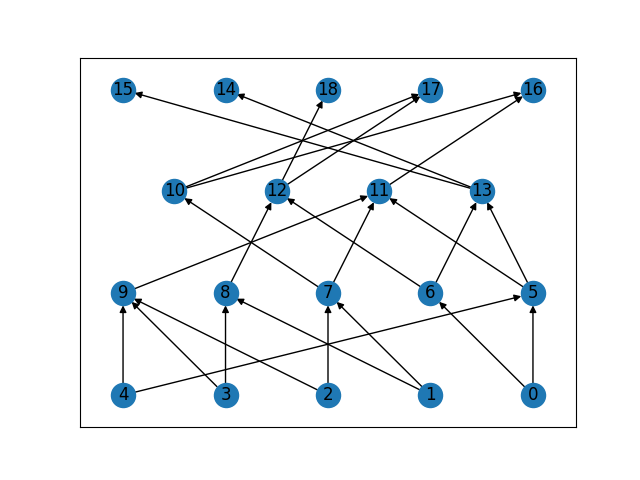
Beta Was this translation helpful? Give feedback.
-
|
(for reference tskit-dev/tskit#852 is the It seems like this representation would work for us if we don't restrict the |
Beta Was this translation helpful? Give feedback.
-
|
Thanks for the insight @jeromekelleher, I've been looking forward to pedigree based simulations in msprime. Having an optional 'k' is a great idea. I was also imagining a single array ( In your example the sample identifiers are ascending integers and therefore the elements of the
So long as your species is dioecious! |
Beta Was this translation helpful? Give feedback.
-
Yes, I think it's consistent with our general design philosophy to refer to objects by their integer IDs. For example, we do this for alleles in the genotypes arrays. Generally, integer indexes are much simpler and more useful than (say) string identifiers when using numpy-based APIs. We can also imagine writing algorithms that work with the pedigree information using numba, where having integer indexes as the primary identifier is much simpler and more efficient than reasoning about string identifiers. Having said that, I totally agree that we would need some way of translating from "external" IDs (like the combined Family ID and Individual ID from plink) to these indexes. I'm imagining that the dataset would contain a string array which contains these "names" which allow us merge on the string IDs. Ideally we'd have some standard array that would correspond to the external ID, but I don't really know what it would be called. What do you think? |
Beta Was this translation helpful? Give feedback.
-
Perfect! Having the indices is a must, but we also need to store the source of those indices. We also need a name for the 'k' dimension in xarray. So far sgkit uses singular for array names and plural for dimension names so I'd suggest:
Or perhaps array Edit: |
Beta Was this translation helpful? Give feedback.
-
|
Here's a rough outline of what I'm imagining for a simple trio: I've used Maybe we should see this as a "sample specification" dataset, more generally, where the pedigree information is just one of the columns we can imagine defining? I.e., filling out the details of this nice picture:
I don't see much point in storing the string parent string IDs - can't we just do Re singular/plural, I think you're probably right that it should be |
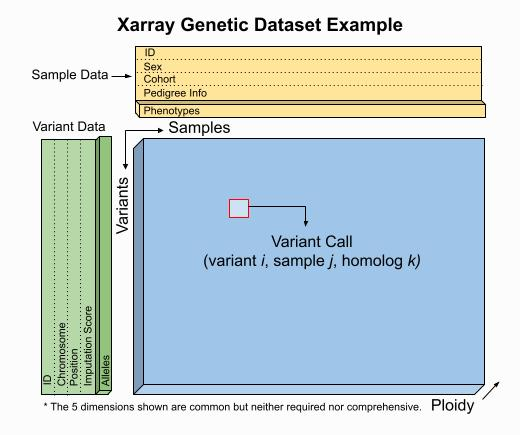
Beta Was this translation helpful? Give feedback.
-
|
The issue I see with this is that the >>> ds["sample_id"] = ["samples"], ["mom", "dad", "child"]
>>> ds["parents"] = ["samples", "parents"], [
... [-1, -1],
... [-1, -1],
... [0, 1]
...]then drop the second sample >>> ds = ds.sel(dict(samples=[True, False, True]))resulting in >>> ds["sample_id"]
["mom", "child"]
>>> ds["parents"]
[
[-1, -1],
[0, 1]
]so the child is now recorded as its own parent. If we repeated that process with a >>> ds["parent_id"]
[
[".", "."],
["mom", "dad"]
]which can be used with >>> ds["parents"]
[
[-1, -1],
[0, -1]
]which reflects that one of the parents is no longer present in the dataset.
Agreed, all columns in a ped file map to the samples dimension. |
Beta Was this translation helpful? Give feedback.
-
Yes, that's a good point. Isn't this also true for variant data though? We don't use string sample IDs to index into the Thinking the workflow through as a combined pedigree+variants dataset is a good idea - what would the subset-of-samples operation look like in both cases? |
Beta Was this translation helpful? Give feedback.
-
This is technically an issue for the relationship between It's actually more likely to be an issue for the
I imagine that any sub-setting would make use of the xarray methods ds2 = ds.sel(dict(
samples=(ds.call_GQ.mean(dim="variants") > 30).values
))More complex operations like sub-setting to the ancestors of an individual within a pedigree would require a function that can generate the indices of ancestor nodes. The simplest way to do this is to generate a networkx DiGraph from the I think the key detail here is storing sufficient data to correct indices after their dimension has been altered. More generally I think we need to clearly document methods that generate arrays of indices and encourage users to run them just prior to their use. This is a fundamental issue with storing indices of dimensions that may change, so it requires some guidelines for best practice. |
Beta Was this translation helpful? Give feedback.
-
|
SGTM @timothymillar, I think we've got a good sketch here of what the requirements are and the API should look like. |
Beta Was this translation helpful? Give feedback.
-
Ah, that's a great point. Could you file an issue? |
Beta Was this translation helpful? Give feedback.
-
|
Done: #795 |
Beta Was this translation helpful? Give feedback.
Uh oh!
There was an error while loading. Please reload this page.
-
Hi all I'd like to start some discussion on the possibility of supporting pedigree data in sgkit.
Background
A pedigree is essentially a DAG recording the familial relationships between individuals. A key aspect of the pedigree DAG structure is that each node typically only descends from two parent nodes in a sexually reproductive species, or from a single node in a clonal species.
There may be a mixture of clonal and sexual reproduction in some breeding systems. For example, some plant varieties have originated as mutant bud-sports (i.e. a single branch/cane exhibiting a novel phenotype due to somatic mutation).
Selfing is a special case of sexual reproduction that may occur in hermaphroditic species in which a sperm and egg cell from a single parent give rise to a new genotype. This differs from asexual reproduction because the sperm and egg cells both represent a random subset of the parent genome. Hence each gamete may contain identical alleles resulting in a potent form of inbreeding.
File formats
Pedigree data is often stored in simple tabular formats where each row represent a single individual/genotype and the parents of each individual are indicated in a "Father" and "Mother" columns (commonly "Sire" and "Dam" in animal breeding).
The rows of these formats are often sorted by topological order (i.e. no individual appears before its ancestors) and this is expected by some software packages.
The ped format is a loose standard which is primarily used for sexually reproducing dioecious diploid species. This is a tabular (white space separated) format used by gatk, plink and many other packages. There are typically six required columns including:
Asexual reproduction is not explicitly supported by the ped format. In some cases a clone is indicated by a single parent (mother or father), however, this may be ambiguous as it can also indicate an unknown parent. Selfing is often represented in ped format by encoding both the father and mother as the parent plant. This is biologically accurate but may be be misinterpreted as asexual reproduction.
The VCF standard supports pedigree descriptions in the header metadata using the
##PEDIGREEtag. This is a very flexible format which allows for explicit labelling of each relationship type and has semi-standard conventions for sexual reproduction ("Father", "Mother") and asexual reproduction ("Original").Basic array representation
The simplest way to store pedigree data in an array structure is a matrix of shape (samples * parental edges) i.e. a row for each sample and a "father" and "mother" column. The values of this matrix may be the parent identifiers or the positional index of each parent based on the sample order.
Pedigree using parent identifiers:
Pedigree using parent indices (
-1indicating unknown):Using the positional indices of parents is more efficient but the indices will be invalidated if the sample order changes. I imagine it would be best to store both forms and have a method to validate/update the indices based upon the labels.
A source of ambiguity in this pedigree structure is when a sample has a single parent. Depending on the species or context, this may indicate a missing parent or that the sample is a clone. A workaround would be to have "father", "mother" and "clone" columns though this may complicate IO of ped files. Another issue with missing parents is that analyses have to deal with "partial" founders instead of having a clear distinction between founders and non-founders.
Pedigree kinship calculations
One of the main use cases for working with pedigree data is to estimate (expected) kinship and inbreeding coefficients based on the pedigree structure. Pedigree based relatedness estimates were pioneered by Pearl (1913) and Wright (1922). Gustave Malécot (1948) developed the coefficient of consanguinity (AKA co-ancestry or kinship) which redefined relatedness in terms of probabilities of alleles being identical by decent (IBD).
Kinship is normally calculated by sorting the pedigree topologically and then recursively calculating coefficients between each new individual based on the relationships of its parents and all preceding nodes. There has been quite a bit of research speeding up kinship estimation in large pedigrees e.g.
Kirkpatrick et al (2019).
One of the big issues with pedigree based kinship estimation methods is that they typically assume that founders are unrelated to one another (i.e. kinship = 0 among all founders). This often leads to underestimates of kinship and inbreeding throughout the pedigree. A means to mitigate this issue is to initialise pedigree based estimation by using marker based estimates among the founding individuals. This results in the expected marker kinship among the descendants. I've read a couple of papers that refer to this approach (e.g. Weir and Goudet (2017)) though Meng et al (2019) is the only paper I know of that go into detail (they handle the x-chromosome in addition to the autosome).
Polyploid and mixed-ploidy data
Kerr et al (2012) and Hamilton and Kerr (2017) generalise pedigree based kinship estimation to auto-polyploids in a very general way which can also handle ploidy manipulation. They introduce the parameters 𝜏 and 𝜆 which respectively indicate the number of genome copies inherited from each parent, and the probability that a pair of alleles in a given gamete descend from a single parental allele (due to a complex phenomenon in in auto-polyploids called double-reduction, or due to gamete manipulations).
The 𝜏 parameter is particularly useful for removing ambiguity in pedigree specifications because the 𝜏 values associated with each parent of a given sample should sum to its ploidy. For example, if a single parent is recorded then 𝜏 makes it clear whether the progeny is a clone (𝜏 = ploidy of sample) or if there is a missing parent (𝜏 < ploidy of sample). In theory this could be generalised to a value per contig (or variant) which would distinguish between autosome and sex chromosomes.
The 𝜆 parameter should theoretically vary across the genome in auto-polyploids. It should increase with distance from the centromeres due to double-reduction.
Issues when joining pedigree and marker data
In my experience there are a few common difficulties that arise when combining pedigree and genotype data into a single dataset. The first is simply joining identifiers which often denote a specific sample for genotype data as opposed to an organism in pedigree data.
A more significant issue is dealing with biological replicates of genotypes as there is no perfect way to represent a replicate in a pedigree graph. The best option is to treat replicates as clones, however, this requires deciding on an "original" replicate which the others are clones of. Generally I try to pick a "best" replicate and remove the others, but comparing pedigree and genetic relatedness is a useful method for picking the "best" replicate ... so it's often a chicken and egg situation.
Another issue is dealing with intermediate pedigree nodes for which there is no genetic sample available. Fortunately, Xarray provides some powerful join operations which enable you to insert a genotype with missing allele data (though this may cause issues downstream).
Basic visualisation
I realise that sgkit is not intended to be a visualisation toolkit however I think it is necessary to provide some basic visualisation of pedigree data (if pedigree data is supported). This is essential to check that the pedigree structure is correct (especially after a join or subset). It is quite easy to encode a pedigree as a DAG using graphviz which is already a dependency of sgkit via dask. A simple
display_pedigreemethod would go a long way towards usability.General graph wrangling
There are some general graph algorithms that are necessary/useful for working with pedigrees. The primary one is topological ordering of pedigree nodes. It's also useful to be able to subset pedigrees based on relationships such as ancestors/descendants. Most of this functionality could be outsourced to networkx.
Beta Was this translation helpful? Give feedback.
All reactions