Replies: 1 comment 6 replies
-
|
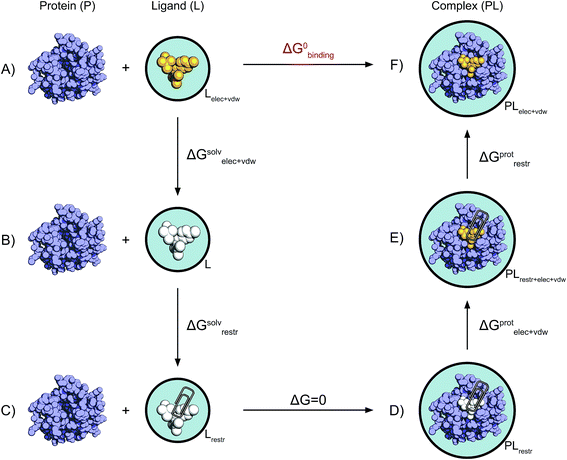
(I moved the issue to a discussion, as I believe it is more suitable here. Feel free to open a new one if you think otherwise.) In principle, using the regular code, no. Because of the way dual-topologies are generated, a molecule must be perturbed into another, so the free energy will be relative. However, I believe
So, the complete answer is: not currently, it will likely be possible in the near future with some tweaking and some (minor?) coding. Furthermore, I think that this would be a quite useful addition to PyAutoFEP and a somewhat low hanging fruit - after the new code allowing VdW+Coulomb perturbation of constant region is deployed. I would be interested in adding it, specially in case there are people interesting in helping code and test. |
Beta Was this translation helpful? Give feedback.
-
|
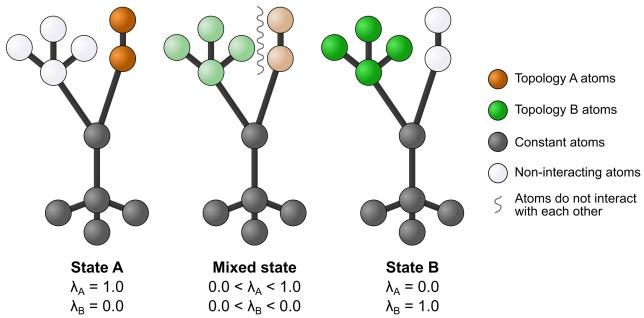
Sorry, I should have unpacked this point. Currently, the terms in the
PS: in case you are asking whether there should be another way to passing a lambda scheme to |
Beta Was this translation helpful? Give feedback.
-
|
Oh, got it. Thank you so much for making things clear. Things seem complicated, but please let me know if any thing I could help you with. |
Beta Was this translation helpful? Give feedback.
-
|
I would love your (and others) help in implementing and/or testing this. I'm opening a discussion about absolute FE implementation after the code for charged perturbations makes it into the master branch. |
Beta Was this translation helpful? Give feedback.
-
|
hello @luancarvalhomartins -- just curious, is this repo now in support of ABFE calculations? |
Beta Was this translation helpful? Give feedback.
-
|
Hi, @spadavec. Thanks for asking. No, unfortunately, not yet. The code to allow perturbing VdW and lambda that I mentioned above is ready but not yet committed. I plan to do that together with the charged-perturbation code, which I wrote but was not able to test extensively yet. After that, it could be possible to implement ABFE as in Chem. Sci., 2016,7, 207-218 with a moderate amount of coding. As I mentioned above, it would be awesome if other were willing to help in coding and/or testing it :) |
Beta Was this translation helpful? Give feedback.
Uh oh!
There was an error while loading. Please reload this page.
Uh oh!
There was an error while loading. Please reload this page.
-
Hi there, thank you for maintaining this awesome repo! I'm wondering whether it is possible to calculate absolute binding free energy with PyAutoFEP, either with the current code base or some modifications on it.
Beta Was this translation helpful? Give feedback.
All reactions