Limitation of Ligand #21
Replies: 8 comments
-
|
Atom order shouldn't matter, so this is almost certainly a bug. The error suggests that the substruct matching failed, and substructure matching is a bit tricky with lone pairs and charges, which is likely whats triggering the bug. I am a bit swamped these day so I wasn't able to find time to test your inputs to understand what maybe going on. I am replying when I'm able to do that. Thanks for the report and sorry for the tardiness. |
Beta Was this translation helpful? Give feedback.
-
|
I tested your inputs and it doesn't seem to be related to atom order, but rather to the fact that your
find_mcs plot using lig13.mol2 find_mcs plot using lig13_new.mol (Note the H in N1033) |
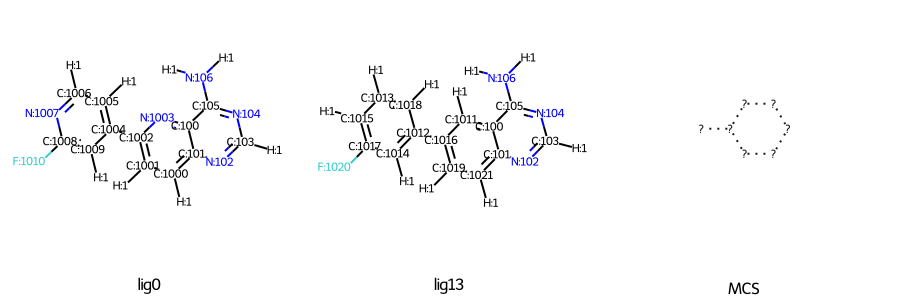
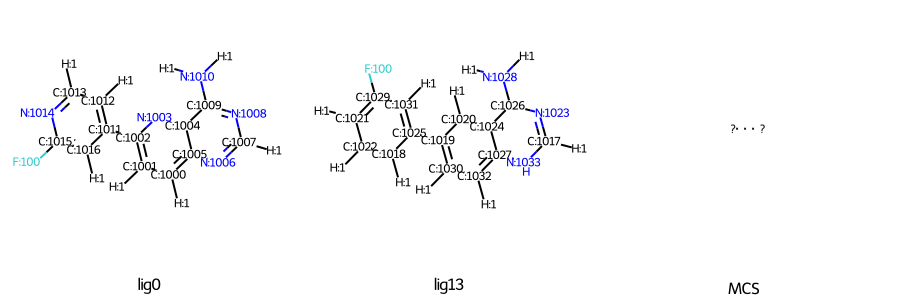
Beta Was this translation helpful? Give feedback.
-
|
Thank you for spending your valuable time on this issue. Another question, do you have any experience with calculations that worked well with periodic structures? |
Beta Was this translation helpful? Give feedback.
-
|
What do you mean exactly by a periodic system? Is the receptor periodic, eg, a microtubule? I have no experience with such a system, maybe using I couldn't figure out what exactly the ligands in your tgz should be, but it seems that something went wrong with the MCS or the topologies. (Should you be working with data you'd rather not share on GitHub, feel free to reach me by email at luancarvalhomartins@gmail.com) |
Beta Was this translation helpful? Give feedback.
-
|
I'm sorry for the late reply. Thank you for your comments. Looking at the input files so far, it seems that the common parts of the ligands should be written in the same order. I would like to try to build the data from scratch again using a well-known system like T4 lysozyme. |
Beta Was this translation helpful? Give feedback.
-
|
I tested fully randomizing ligand atom order for a test system and the MCS and topologies were correct, regardless of the atom order. This matches what I expected from the code. During dual topology generation, the ligand A atom order will be used as reference and part B atoms will be added after the last atom of constant or part A, which one is the last. Did you observe a problem for a specific system? Regarding T4 lyso, I sometimes use PDBID TLX6 as a test receptor. |
Beta Was this translation helpful? Give feedback.
-
|
I'm sorry for the late reply. Thank you for sharing the information about not having to worry about the atom order. |
Beta Was this translation helpful? Give feedback.
-
|
I don't know what is causing the issue you obserserved, as I would need the original ligand and topology files. I'm moving this issue to a discussion. In case you need any help, feel free to reply, open another issue, or reach me by email. |
Beta Was this translation helpful? Give feedback.
Uh oh!
There was an error while loading. Please reload this page.
-
I'm currently creating input for another system.
I have a few questions about Ligand and I would like to share them with you.
I have found that some input files can be created with the same structure, but some cannot, depending on the order of the atoms. Is there anything I should pay attention to when preparing the files?
Success LIG13.mol -> LIG0.mol
Failure LIG13_new.mol→LIG0_new.mol
https://github.com/baba-hashimoto/PyAutoFEP/tree/master/files
[ERROR] Less than 3 common atoms were found between molecule LIG13 (SMILES=<rdkit.Chem.rdchem.Mol object at 0x14562bb603a0>) and LIG0 (SMILES=(17, 17))
More than half of the constituent atoms belong to dual_topology, have you ever calculated such a system? In this case, I was able to prepare the input file, but I got an error in the energy minimization calculation.
[ exclusions ] ; Added by dual_topology
11 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 ; Added by dual topology
12 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 ; Added by dual topology
Beta Was this translation helpful? Give feedback.
All reactions