Replies: 3 comments 30 replies
-
|
Can you explain what you mean by "% error expression"? |
Beta Was this translation helpful? Give feedback.
-
|
So I should just ignore any TargetID without an identifier? (for example I would delete the first three rows in this sample data set) |
Beta Was this translation helpful? Give feedback.
-
|
You keep every Target ID in the table I provided |
Beta Was this translation helpful? Give feedback.
-
|
In that case 95% of the data set is NA, is that normal? |
Beta Was this translation helpful? Give feedback.
-
|
You want to know the expression pattern of every Target ID, including those that are not expressed at all. |
Beta Was this translation helpful? Give feedback.
-
|
so you really have a line like line 1? no target ID? |
Beta Was this translation helpful? Give feedback.
-
|
I think I see the problem You should be using left_join (or something similar) where you retain only annotation information that matches data table with expression |
Beta Was this translation helpful? Give feedback.
-
|
|
Beta Was this translation helpful? Give feedback.
-
|
we do - but your line 1 has no count data. |
Beta Was this translation helpful? Give feedback.
-
|
left join would not remove line 2 and 3. |
Beta Was this translation helpful? Give feedback.
-
Beta Was this translation helpful? Give feedback.
-
|
Okay I figured it out, I did need to add a left.join at line 42 of the code I posted above, thanks! |
Beta Was this translation helpful? Give feedback.
-
|
Put a link to the GitHub repo where you are working.. that would be easiest way to understand and troubleshoot |
Beta Was this translation helpful? Give feedback.
-
|
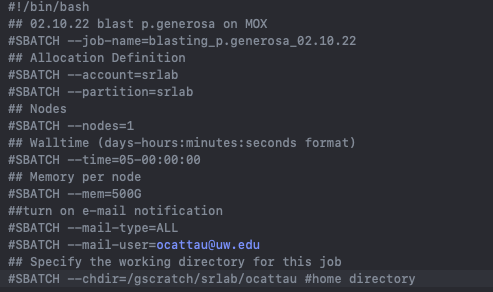
what does you slurm header look like? |
Beta Was this translation helpful? Give feedback.
-
|
|
Beta Was this translation helpful? Give feedback.
-
|
I think I need to wait till Sam's job is done because of memory issues |
Beta Was this translation helpful? Give feedback.
-
|
change mem to 100G |
Beta Was this translation helpful? Give feedback.
-
|
Full explanation - we have two nodes Sam is using the 500M, but you do not require that. When you set mem to <120G your job will start on 2nd node. |
Beta Was this translation helpful? Give feedback.
Uh oh!
There was an error while loading. Please reload this page.
Uh oh!
There was an error while loading. Please reload this page.
-
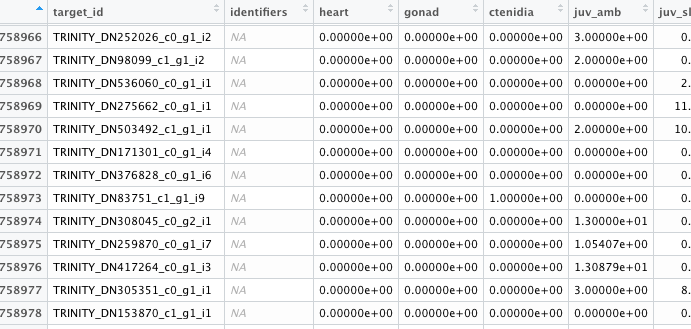
Lets say my data looks like this table after the count data has been aligned with the blast data. In order to calculate % error expression, my first reaction would be to calculate % NA in the Identifier column, but sometimes there is count data without an identifier.
My second guess would be to filter the tpm so only values > 0 are counted, then calculate the % that has any value at all, but would some values be NA and some 0?
I don't think it makes any sense to calculate % error expressed using the Target_ID column since most of them have values but sometimes there are identifiers that don't have a corresponding Target_ID.
If I use the R function complete.cases, it removed any row with NAs which would delete all rows 1-3 in this example, and in my mind if there is count data but no identifiers, wouldn't that mean the gene exists but we haven't identified it yet? Any help with this data wrangling would be appreciated.
Beta Was this translation helpful? Give feedback.
All reactions