-
|
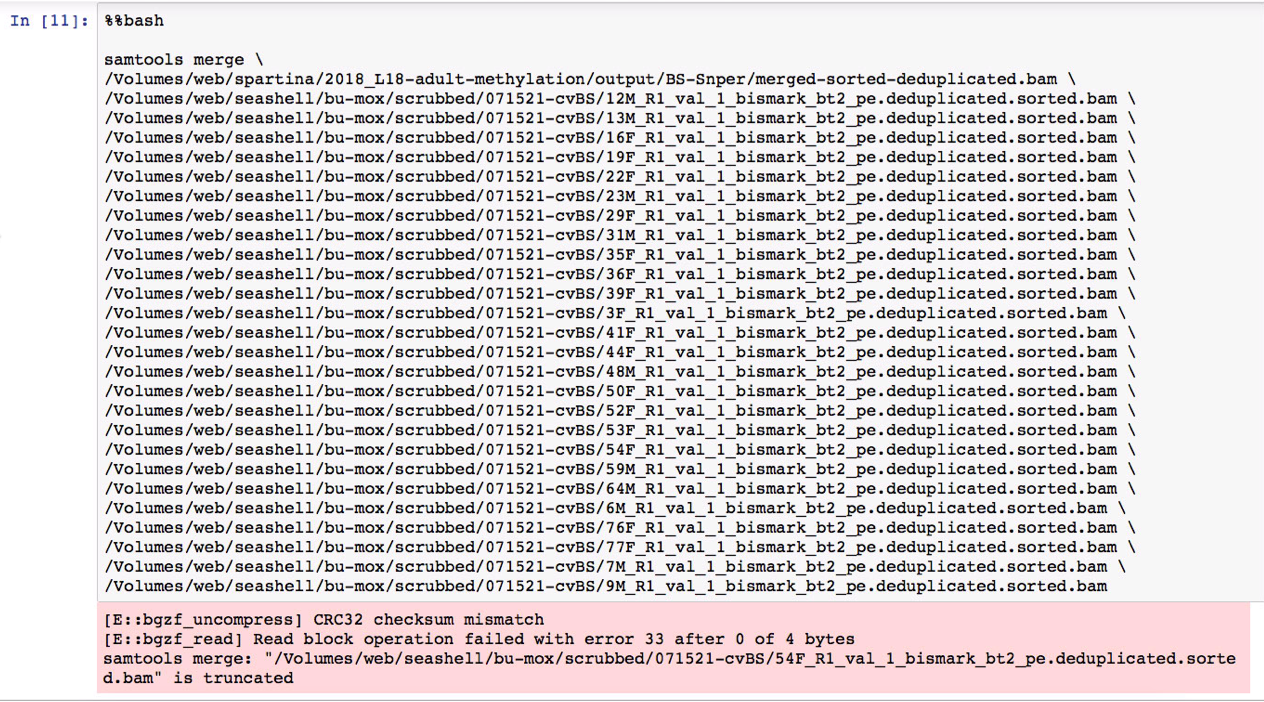
@sr320 I'm using the deduplicated and sorted BAM files you generated for the Cv gonad samples (saved here). I need to merge all the files before calling SNPs. Tried doing so in this Jupyter notebook but got the following error:
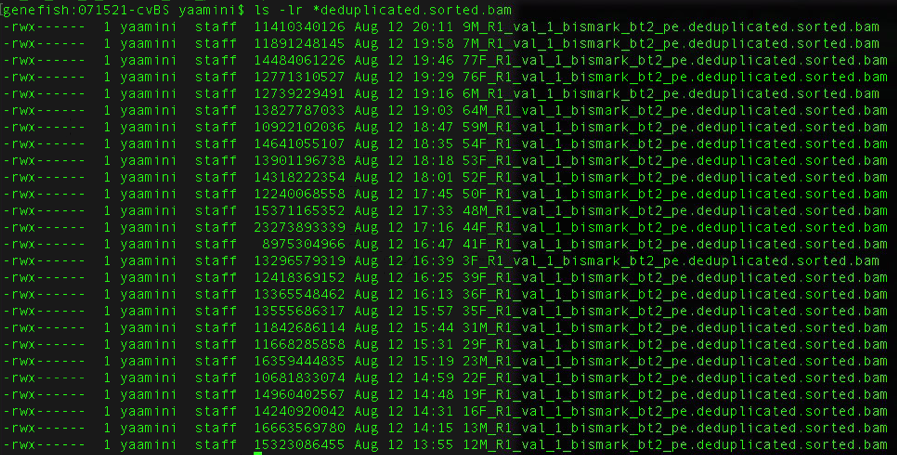
Is there a reason why I'm getting this error/how would I check if this file is actually truncated? File sizes for reference: edited to include the correct second screenshot and notebook link. |
Beta Was this translation helpful? Give feedback.
Replies: 2 comments 2 replies
-
|
A few things:
|
Beta Was this translation helpful? Give feedback.
-
|
Beta Was this translation helpful? Give feedback.
-
|
Ah! You will not (and should not) be able to work this way. The files are too large to transfer over the internet while processing them. Also, processing files this way is MASSIVELY slower than running things locally, due to the data transfer requirements back and forth, combined with the slower processor of Gannet having to handle that stuff simultaneously. |
Beta Was this translation helpful? Give feedback.
-
|
In regards to Number 2, memory (RAM) and hard drive (HDD) are different things. The memory to run this is still based off of what amount of RAM your computer has (since this is what's running the commands). |
Beta Was this translation helpful? Give feedback.
Ah! You will not (and should not) be able to work this way. The files are too large to transfer over the internet while processing them. Also, processing files this way is MASSIVELY slower than running things locally, due to the data transfer requirements back and forth, combined with the slower processor of Gannet having to handle that stuff simultaneously.